In previous post, we discussed some of the introduction to Large Language Models (LLMs) and how they are constructed, trained, and utilized. Beginning with this post in the Biomedical LLMs series, we will explore their applications in biomedical domains. This post will concentrate on a few LLMs for genomics (e.g. DNA and RNA).
DNA Language Models
DNABERT
DNABERT (Ji et al., 2021) is designed to encoder genomic DNA sequences by adapting the Bidirectional Encoder Representations from Transformers (BERT) model. DNABERT utilizes a Transformer’s encoder architecture characterized by attention mechanisms, which effectively capture both local and long-range dependencies in DNA sequences and offer contextual representation of the input DNA sequences. The encoder-only architecture is identical to the BERT base model, comprising 12 transformer layers, each with 768 hidden units and 12 attention heads.
DNABERT employs a k-mer tokenization strategy to segment DNA sequences into overlapping k-mers, which provides a more coarse-grained and computationally cheaper way to capture the contextual information than base-by-base tokenization. The authors experimented with four k-mer sizes: 3, 4, 5, and 6. Each k-mer model (DNABERT-3, DNABERT-4, DNABERT-5, DNABERT-6) has a vocabulary size of 4^k+5 tokens, including special tokens such as [CLS] (classification), [PAD] (padding), [UNK] (unknown), [SEP] (separator), and [MASK] (masking). Each version captures different levels of contextual information, with DNABERT-6 generally performing the best due to its richer context representation. Moreover, learned positional embeddings (likely learns a simple lookup table via a linear layer) were used to add to the token embeddings.
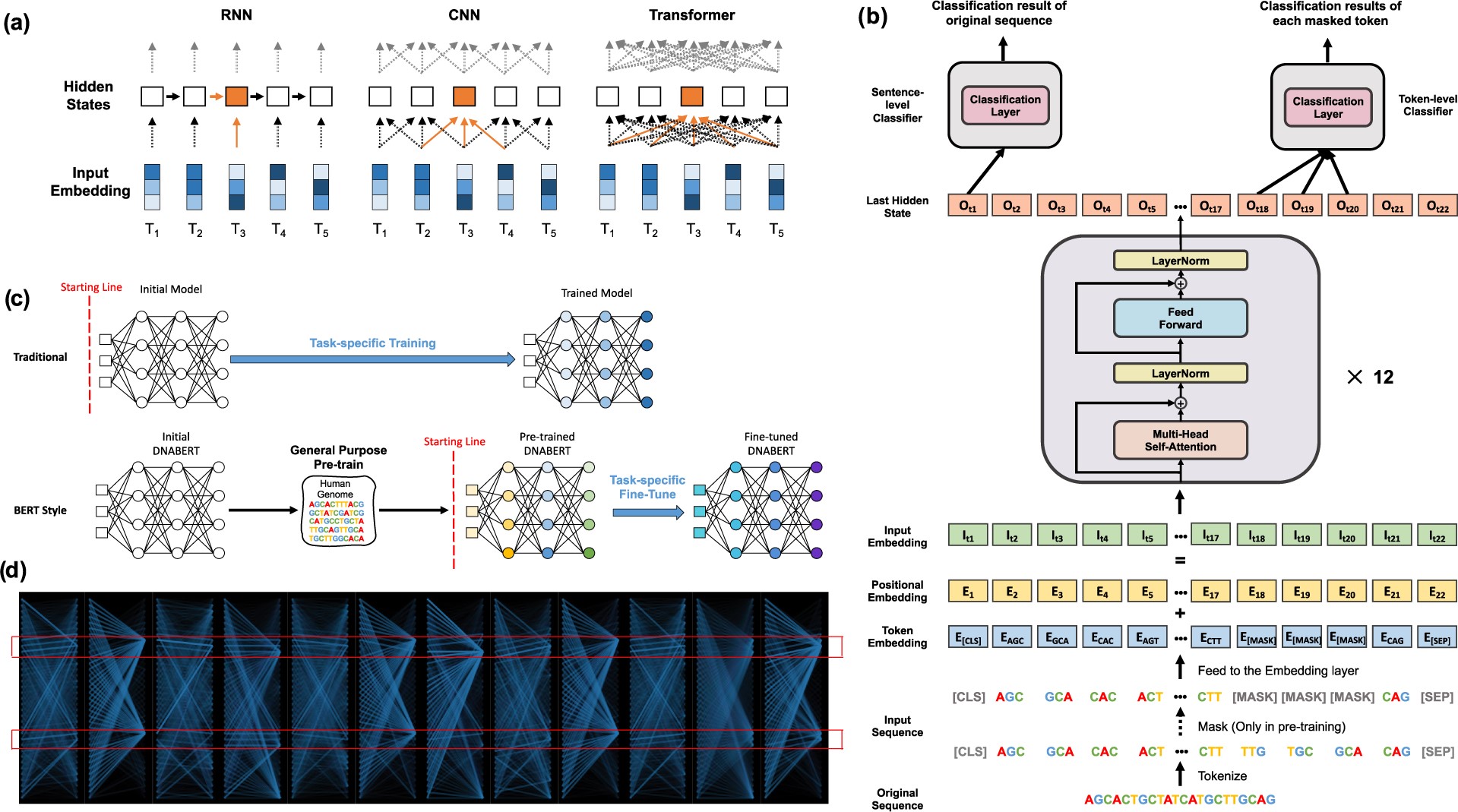
Fig 1. Architecture and Key Features of the DNABERT Model. (a) Contextual understanding in RNN, CNN, and Transformer models: Comparison of how each model processes input tokens (T1-T5) to generate hidden states. RNNs use sequential information flow, CNNs focus on local context, while Transformers employ global self-attention mechanisms. (b) DNABERT’s structure: The model inputs tokenized k-mer sequences, including special tokens (CLS, SEP, MASK). This data passes through an embedding layer and 12 Transformer blocks. The model produces sentence-level classifications from the first output and token-level classifications from individual masked token outputs. (c) DNABERT’s versatility: The model uses general pre-training, allowing fine-tuning for various specific tasks with appropriate data. (d) DNABERT’s attention visualization: An example showing global attention patterns across 12 attention heads, demonstrating the model’s ability to focus on significant regions, such as known binding sites within a sequence.. The figure is Fig.1 from Ji et al., 2021.
The pretraining dataset for DNABERT was derived from the human genome, which has 3 billion bases. Sequences were generated through direct non-overlapping splitting and random sampling, with lengths varying between 5 and 510 bases. The pretraining process involved masking 15% of k-mers in the sequences for the first 100,000 steps and increasing this to 20% for the final 20,000 steps. The model was pretrained for 120,000 steps with a batch size of 2,000. The training process for DNABERT took approximately 25 days on 8 NVIDIA 2080Ti GPUs.
DNABERT exhibits versatility across various genomic sequence-related tasks. It demonstrated state-of-the-art (SOTA) performance in 2021 for predicting promoters, splice sites, and transcription factor binding sites. By comparing the fine-tuned DNABERT pre-trained on the human genome with 78 mouse ENCODE ChIP-seq datasets against CNN, CNN+LSTM, CNN+GRU, and randomly initialized DNABERT models on cross-genome sequence prediction tasks, the authors show that the pre-trained DNABERT significantly outperforms all baselines. This suggests that pre-training not only enhances model performance by providing a better starting point for training but also enables robust transfer across species, even with considerable dissimilarities in non-coding regions, highlighting the model’s ability to capture shared deep semantic features across genomes. Furthermore, by visualizing the attention maps, DNABERT provides insights into the relative importance of different nucleotides and their semantic relationships within sequences. This interpretability demonstrates that deep learning models need not be “black boxes” - the model’s attention patterns can help identify conserved sequence motifs and predict functional genetic variants by highlighting biologically meaningful patterns in the data.
DNABERT-2
DNABERT-2 (Zhou et al., 2023) is a refined genome foundation model designed to improve upon the limitations of its predecessor, DNABERT. DNABERT-2 is built upon the Transformer’s encoder architecture, similar to BERT and DNABERT, but incorporates a few key changes:
- DNABERT-2 adapts SentencePiece 1 (Kudo, 2018) with Byte Pair Encoding (BPE) 2 (Sennrich et al., 2015) to tokenize DNA sequences instead of using k-mer tokenization. BPE iteratively merges the most frequent co-occurring genome segments. This method overcomes the limitations of k-mer tokenization, such as information leakage 3 and poor computational efficiency, by providing a more streamlined and effective approach to sequence representation. This switch addresses computational inefficiencies and sample inefficiencies associated with k-mer tokenization. 8 vocabularies with target sizes ranging from 2^8 to 2^15 were tested for the BPE tokenization strategy. The best performing model was trained with a vocabulary size of 2^15, where larger vocabulary size means longer sequences but more sparse updates to the embedding layer. Evaluating different vocabulary sizes on a multi-species genome dataset on the Genome Understanding Evaluation (GUE) benchmark led the authors to use a vocabulary size of 2^12 = 4096 for training the final DNABERT-2 model.
- Except for replacing k-mer tokenization with BPE for better tokenization efficiency and sequence representation, DNABERT-2 abandons learned positional embeddings by integrating Attention with Linear Biases (ALiBi) (Press, et al, 2021) to overcome input length limitations. Unlike approaches that add positional embeddings to word embeddings, ALiBi biases query-key attention scores with a penalty proportional to the distance between tokens, enabling the model to extrapolate efficiently to sequences longer than those encountered during training. Additionally, DNABERT-2 also employs Flash Attention (Dao, et al., 2022) and Low Precision Layer Normalization to boost computational efficiency. This model comprises 117M parameters, making it substantially smaller yet more efficient than other models in its class.
The pre-training of DNABERT-2 involved two main datasets: a human genome dataset consisting of 2.75 billion nucleotide bases, and a multi-species genome dataset comprising genomes from 135 species, totaling 32.49 billion nucleotide bases. This multi-species dataset ensures a broader representation and diversity of genetic sequences. Maximum sequence length was set to 510bps4 for DNABERT.
The pre-training of DNABERT-2 took approximately 14 days using eight Nvidia RTX 2080Ti GPUs. The training process employed a batch size of 4096 and a maximum sequence length of 128, optimized using the AdamW optimizer with specific hyperparameters.
As briefly mentioned earlier, DNABERT-2 introduces the Genome Understanding Evaluation (GUE) benchmark to evaluate DNEBERT-2. GUE includes 36 datasets across nine genome analysis tasks from four species. This benchmark enables a standardized assessment of model performance across various tasks such as promoter detection, transcription factor prediction, and species classification. While the input size of datasets in GUE ranges from 5000 to 10000, the authors splitted the input into 512bps segments and use the averaged embedding of each segment as the input to the model during their benchmarking. Also, the DNABERT-2 model is pre-trained purely on 700-bps sequences. Thanks to the extrapolation capability offered by ALiBi, the authors showed that such a setting still performs well even on 10000-bps sequences with a few epochs of fine-tuning.
Compared to its predecessor DNABERT, DNABERT-2 achieves superior performance with significantly reduced computational cost and model size. In the authors evaluation, it performs on par with “SOTA” Nucleotide Transformers in 2023 while being 21 times smaller and requiring 92 times less GPU time for pre-training. This efficiency makes DNABERT-2 particularly suitable for fine-tuning on consumer GPUs.
Nucleotide Transformer
The Nucleotide Transformer (NT) is a foundation model developed by InstaDeep in collaboration with Nvidia, designed for encoding genomic sequences (Dalla-Torre, et al., 2023). This model series, ranging from 50M to 2.5B parameters, has been developed over recent years with continuous improvements in training techniques and architectural enhancements.

Fig 2. Overview of the Nucleotide Transformer: pre-training, fine-tuning, analysis, and comparison of foundational models for genomics. (a,b) Illustration of the Nucleotide Transformer’s training process (a) and its application to downstream genomic prediction tasks via fine-tuning (b). Probing for downstream tasks follows a similar approach but excludes rescaling weights. (c) Comparative analysis of the Nucleotide Transformer models against other foundational genomics models, focusing on receptive field size, number of parameters, and performance across a benchmark comprising 18 curated downstream tasks. (d) Visualization of genomic features considered in downstream tasks. The figure is Fig.1 from Dalla-Torre, et al., 2023.
NTs employ an encoder-only transformer architecture similar to BERT. They use non-overlapping 6-mer DNA tokens as a trade-off between sequence length (up to 6kb) and embedding size, achieving the highest performance compared to other token lengths. NT-v1 models use a learnable positional encoding layer that accepts a maximum of 1000 tokens 5. Each model includes transformer layers with layer normalization, multi-head self-attention, and a two-layer perceptron with GELU activations. The parameter sizes for NT-v1 models range from 500M to 2.5B.
The updated NT-v2 models feature architectural advancements such as rotary embeddings (Su, et al., 2024)6 and swiGLU activation (Shazeer, 2020). They eliminate MLP biases and dropout mechanisms to improve efficiency. NT-v2 models extend the context length to 12kb by accepting up to 2048 tokens and include variants with parameter sizes from 50M to 500M.
NT models are pre-trained on diverse datasets to ensure comprehensive genomic representation:
- Human Reference Genome: Based on the GRCh38/hg38 assembly, encompassing 3.2 billion nucleotides.
- 1000 Genomes Project (1000G): Comprising 3,202 high-coverage human genomes, representing 20.5 trillion nucleotides from 27 geographically diverse populations.
- Multispecies Dataset: Includes 850 genomes from various species, totaling 174 billion nucleotides, selected to maximize diversity and functional relevance across different phyla.
The tokenization process converts nucleotide sequences into 6-mer tokens, with a vocabulary of 4104 tokens, including special tokens for padding [pad], masking [mask], and sequence start [CLS]. All nucleotides other than A, T, C, G were replaced by N before tokenization. The models are trained using a masked language modeling (MLM) approach similar to BERT, where 15% of the tokens in each sequence are masked, and the model learns to predict these masked tokens. Training utilizes the Adam optimizer with a learning rate schedule and gradient accumulation to handle large batch sizes effectively, with an effective batch size of 1M tokens per batch. Training is conducted on the Cambridge-1 Nvidia supercomputer, using 128 A100 GPUs across 16 nodes. NT-v1 models require up to 28 days for training, while NT-v2 models are trained for extended durations, with the largest models processing up to 1 trillion tokens to better understand the scaling laws (Kaplan, et al., 2020).
NT models are evaluated based on their performance across various genomic tasks:
-
Scaling Laws: NT models, ranging from 50M to 2.5B parameters, were trained on extensive datasets. Larger models captured more complex genomic patterns, resulting in better generalization and accuracy. Architectural advancements in NT-v2 allowed smaller models to achieve results comparable to larger ones. These findings emphasize the importance of optimizing training techniques and extending context lengths to enhance model performance.
-
Benchmarking: NT models were tested on 18 diverse genomic tasks, such as predicting epigenetic marks, chromatin profiles, and splice sites. Larger models consistently outperformed smaller ones, with the 2.5B parameter NT-v2 model showing superior accuracy in most tasks. The NT-v2 500M parameter model achieved similar performance to the 2.5B parameter model due to architectural improvements and longer training durations. Smaller models, like the 50M and 100M parameter versions, also performed well in less complex tasks or resource-limited environments. This benchmarking highlighted the trade-off between model size, computational efficiency, and task complexity.
-
Model Selection: Selecting NT models depends on the specific downstream tasks. For tasks like predicting epigenetic marks and chromatin profiles, larger models (500M-2.5B parameters) are recommended due to their higher accuracy and ability to capture intricate dependencies. For splice site prediction, the NT-v2 500M model offers excellent performance comparable to SOTA models. For simpler tasks or when computational resources are limited, smaller models (50M-100M parameters) provide sufficient performance and efficiency. Understanding the specific requirements and constraints of the application is crucial in selecting the most appropriate NT model.
The NT models have shown significant improvements over existing benchmarks like DNABERT and Enformer (see below), particularly in tasks involving human genomic data.
Enformer
Enformer (Avsec, et al., 2021), developed by a team at DeepMind in collaboration with Calico Life Sciences and Google, leverages transformer architecture to predict gene expression and chromatin states from DNA sequences in humans and mice. While Enformer does not involve pre-training tasks like other large language models (LLMs), its adoption of a BERT-like architecture makes it highly relevant to this post.

Fig 3. Overview of the Enformer architecture. The model is trained to predict human and mouse genomic tracks at a 128-bp resolution using 200 kb of input DNA sequence. It replaces dilated convolutions with transformer modules, expanding its receptive field fivefold to detect elements up to 100 kb away, compared to 20 kb in Basenji2. Detailed architecture settings can be found in Extended Data Fig. 1 of the original paper (Avsec, et al., 2021), with a comparison to Basenji2. The figure was originally Fig. 1a but is linked through DeepMind’s corresponding blog post.
Enformer combines deep convolutional neural networks with transformer blocks, significantly extending its receptive field. The model processes 196,608 bps of DNA sequence input and predicts 5,313 genomic tracks for humans and 1,643 for mice. It consists of seven convolutional layers followed by eleven transformer layers, using attention mechanisms to integrate information from distal genomic elements up to 100 kb away. This setup contrasts with previous SOTA models like Basenji2, which can only integrate information from up to 20 kb away.
Regarding tokenization, input DNA sequences are one-hot encoded, with each nucleotide represented by a unique vector (A = [1,0,0,0], C = [0,1,0,0], G = [0,0,1,0], T = [0,0,0,1]). This encoding feeds into the convolutional layers, which reduce the spatial dimension, allowing the transformer layers to capture long-range interactions effectively. Moreover, the model employs custom relative positional encodings like Transformer-XL paper does (in short that is to add relative positional encodings $R_{ij}$ to the $q_i k_j^T$ ) to enhance its ability to distinguish between proximal and distal regulatory elements and to differentiate positions upstream and downstream of the transcription start site (TSS). This approach ensures effective integration of long-range genomic interactions, crucial for accurate gene expression prediction.
Enformer was trained using a multitask learning framework on a vast dataset encompassing most of the human and mouse genomes. The training involved 34,021 human and 29,295 mouse sequences, with additional validation and test sets. The dataset included various genomic assays such as transcription factor (TF) chromatin immunoprecipitation and sequencing (ChIP-seq), histone modification ChIP-seq, DNase-seq, and ATAC-seq, providing a comprehensive set of genomic tracks.
Training was conducted on 64 TPU v3 cores over approximately three days, with optimization handled by the Adam optimizer. The model’s training and validation employed Poisson negative log-likelihood loss (same as Basenji2 did) 7, and data augmentation techniques like random shifting and reverse-complementing the input sequences were used to enhance robustness.
Enformer demonstrated superior performance in gene expression prediction compared to Basenji2, with mean correlation improvements from 0.81 to 0.85 in predicting RNA expression at TSSs of human protein-coding genes. The model also showed enhanced ability to predict tissue-specific gene expression and the effects of genetic mutations on gene expression, validated by CRISPR interference assays and population eQTL studies.
Although Enformer is not an LLM, its generic architecture and trained model make it versatile for various applications, including fine-mapping of human disease associations, understanding cis-regulatory evolution, and potentially designing synthetic enhancers for specific cell types. Its ability to predict regulatory activity from DNA sequence alone presents a significant advantage in genomic research.
Despite its advancements, the authors acknowledged in their paper that Enformer is limited by its reliance on the cell types and assays present in its training data. It cannot generalize to new cell types or assays not included in its training set. Future improvements could involve integrating 3D genome organization data to better model genomic interactions and expanding training datasets to include more cell types and organisms. Additionally, advancements in computational efficiency and hardware could further enhance the model’s scalability and performance. Moreover, other studies suggest that Enformer may not present SOTA results in benchmarking. Methods like nucleotide transformers we mentioned previously can outperform Enformer although Enformer has a much larger receptive field.
GPN
The Genomic Pre-trained Network (GPN) is a language model designed for genome-wide variant effect prediction, leveraging unsupervised pretraining on genomic DNA sequences (Benegas, et al., 2023). It is particularly notable for its application to predicting the functional impact of genetic variants in Arabidopsis thaliana, a model organism for plant biology.
GPN is based on a customized transformer-encoder where the traditional multi-head attention layers are replaced by convolutions. This design leverages the efficiency of convolutional networks in capturing local dependencies, which are prevalent in genomic sequences. The core of GPN consists of 25 convolutional blocks, each incorporating a dilated convolutional layer followed by a feed-forward layer, with intermediate residual connections and layer normalization. The model maintains a fixed embedding dimension of 512 across all layers. This architecture allows GPN to effectively model both local and long-range dependencies within genomic sequences. GPN was trained for 150,000 steps over four days using four NVIDIA A100 80 GB GPUs.

Fig 4. Overview of the Genomic Pre-trained Network. GPN predicts nucleotides at masked positions in a 512-bp DNA sequence. During training, 15% of positions are masked, while only the variant position is masked during variant effect prediction. The model uses a convolutional neural network to generate embeddings for each position and outputs nucleotide probabilities for masked positions. Training is performed with cross-entropy loss on the reference sequence, and the variant effect prediction score is the log-likelihood ratio between the alternate and reference alleles. Here, L represents the window length in base pairs, D denotes the embedding dimension, REF is the reference allele, and ALT is the alternate allele. The figure was Fig. 1 from Benegas, et al.,2023.
Unlike models that utilize k-mers or byte-pair encoding for tokenization, GPN uses single-nucleotide tokens. This approach simplifies the interpretation of model outputs, which is crucial for variant effect prediction. The model does not incorporate explicit positional embeddings; instead, it leverages the convolutional layers’ structure to capture positional information within the sequence. This design introduces translational equivariance as an inductive bias through the convolutional operations. While this assumption can be advantageous, it may also limit generalization when this bias does not align with the data.
GPN was pretrained on unaligned reference genomes from Arabidopsis thaliana and seven related species within the Brassicales order. The training dataset included various genomic regions such as exons, promoters, and random genomic windows, ensuring a comprehensive representation of the genome. The model processes input DNA sequences of 512 base pairs, where 15% of the positions are masked during training. The goal is to predict the nucleotides at these masked positions, facilitating the learning of complex genomic features and structures. To address the overrepresentation of repetitive elements, the training loss was adjusted to down-weight these regions, improving the model’s performance on non-repetitive, functionally significant regions.
GPN is designed to predict the effects of genetic variants across the genome, making it a powerful tool for genome-wide association studies (GWAS) and fine-mapping of causal variants. It outperforms traditional conservation scores like phyloP and phastCons in predicting variant effects in Arabidopsis thaliana.
The authors highlight that GPN’s ability to learn and predict gene structures and DNA motifs without any supervision. This capability is crucial for identifying transcription factor binding sites and other regulatory elements in the genome. Additionally, GPN’s predictions show a strong correlation with functional genomic regions, as evidenced by its high accuracy in distinguishing coding sequences, untranslated regions, and introns. This work should remind us that conventional CNN-based models can also work well sometimes.
HyenaDNA

Fig 5. Overview of HyenaDNA. The figure was from the GitHub profile page of the author’s repo for this work (link).
HyenaDNA is a genomic foundation model designed to handle long-range dependencies in DNA sequences at single nucleotide resolution (Nguyen, et al., 2024). HyenaDNA addresses the limitations of previous Transformer-based genomic models that were constrained by the quadratic scaling of attention mechanisms. These earlier models could only handle contexts of up to 4k tokens, significantly limiting their ability to model long-range interactions in genomic sequences.
HyenaDNA is based on the Hyena operator (Fig. 6), a convolutional model that can process long contexts with sub-quadratic time complexity. This architecture allows HyenaDNA to scale linearly with sequence length, enabling the modeling of up to 1 million tokens in a single context. The key components of the HyenaDNA architecture include:
-
Implicit Convolutions: The Hyena operator uses long convolutions parameterized by a neural network, which are evaluated using Fast Fourier Transform (FFT) for efficient computation.
-
Element-wise Gating: These gates modulate the input based on learned parameters, allowing the model to apply context-specific operations at each token position.
-
Single Nucleotide Tokenization: Unlike previous models that relied on k-mers or other aggregation techniques, HyenaDNA tokenizes DNA sequences at the single nucleotide level, preserving fine-grained information crucial for understanding genetic variations.

Fig 6. HyenaDNA Block. (left) The HyenaDNA block resembles a Transformer-decoder block, but with the attention mechanism replaced by a Hyena Operator. (middle) The Hyena Operator integrates long convolutions with element-wise gates, where the gates are derived from projections of the input using dense and short convolutional layers. (right) The long convolutions are parameterized implicitly through an MLP, which generates the weights for the long Hyena filters. The figure was from (https://hazyresearch.stanford.edu/blog/2023-06-29-hyena-dna).
HyenaDNA is a decoder-only model that was pretrained on the human reference genome using next-nucleotide prediction. This autoregressive training employed a sequence length warm-up technique, gradually increasing the context length to stabilize training and improve efficiency. The model demonstrated significant improvements in perplexity with longer contexts, indicating better prediction accuracy. The pretraining used a minimal DNA vocabulary consisting of ‘A’, ‘C’, ‘G’, ‘T’, and special tokens.
HyenaDNA was pretrained using Nvidia A100 GPUs (exact number was not disclosed by authors), with training times varying based on sequence length and model size. For example, training a model with a context length of 1 million tokens took approximately 4 weeks. The model sizes used in training ranged from 400k to 6.6M parameters, and it employed gradient checkpointing to manage memory usage efficiently during training.
Despite being as an auto-regressive, decoder-only model for predicting the next nucleotide, HyenaDNA can be used in discriminative tasks such as classification. This adaptability is achieved through techniques like sequence-level pooling and soft prompting.
-
In sequence-level pooling, the outputs of HyenaDNA are aggregated across the genomic sequence to form a cohesive representation, which can then be funneled through a classification head to predict labels for the entire sequence. This method leverages the contextual information encoded by the model to classify sequences based on their entire compositional makeup.
-
For discriminative tasks, HyenaDNA can also leverage soft prompting (check relevant information in the first post of this series), which involves integrating trainable tokens directly into the input sequence. These tokens are optimized during training to adapt the model’s focus towards relevant features for the classification task, enabling effective in-context learning without extensive retraining of the model. This method seems elegant as it allows for flexible adaptation to new tasks by modifying only a small part of the model’s input, making HyenaDNA a powerful tool for genomic classifications and other discriminative tasks, while preserving computational efficiency and model integrity.
In authors’ evaluation, HyenaDNA excels in various genomic tasks, including species classification and regulatory element identification. HyenaDNA achieved SOTA performance on 12 out of 18 benchmarks from the Nucleotide Transformer dataset, despite using significantly fewer parameters and pretraining data. On the GenomicBenchmarks dataset, HyenaDNA surpassed the SOTA on 7 out of 8 datasets, with notable improvements in enhancer identification.
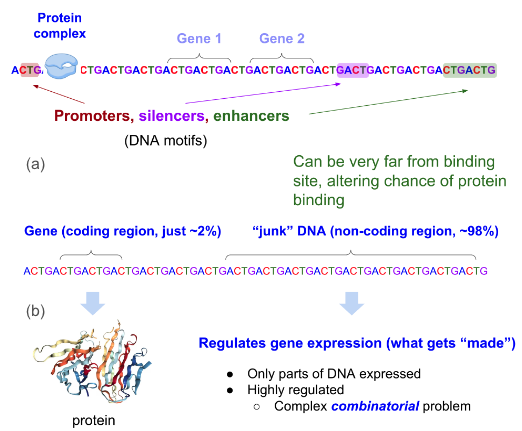
Fig 7. Why single nuceotide resolution and long context are important. (a) DNA motifs play a crucial role in gene regulation. Variants in regulatory regions such as promoters, silencers, and enhancers influence the binding of transcription factors and RNA polymerases to DNA, ultimately affecting gene expression. (b) Single-nucleotide resolution is essential for precise variant analysis, but not all DNA sequences are relevant to gene expression. Relevant sequences may be located far apart within the genome, necessitating a long contextual span to capture their interactions effectively. The figure was adapted from (https://hazyresearch.stanford.edu/blog/2023-06-29-hyena-dna).
HyenaDNA offers significant advancements in genomic sequence modeling due to its ability to handle long-range contexts of up to 1 million tokens, which allows it to capture intricate dependencies across vast genomic sequences. This capability is particularly beneficial for tasks that require understanding long-range interactions, such as regulatory element identification and species classification. Additionally, the model’s use of single nucleotide tokenization ensures that every subtle genetic variation is preserved, enabling precise detection of single nucleotide polymorphisms (SNPs) and mutations. Check Fig.7 to better understand such biological significance further. Moreoever, the architecture, based on implicit convolutions, provides efficient computation with sub-quadratic scaling, making it more effective and faster than traditional Transformer-based models. This efficiency is further enhanced by techniques like gradient checkpointing and sequence length warm-up during training.
Despite its innovations, the pretraining of HyenaDNA was conducted on a single human reference genome, which may limit the model’s generalizability to broader genomic datasets or to genomes from different species. Incorporating multiple genomes in the training process could enhance its robustness and applicability. Expanding the model’s application beyond DNA sequences to other biological sequences, such as proteins and RNA, also presents an area for future research and potential enhancement.
Evo
Evo is a cutting-edge DNA foundation model that generalizes to prediction tasks and generative design at scales ranging from molecular to whole genomes (Nguyen, et al., 2024). Developed by key contributors of HyenaDNA in collaboration with Stanford University, Arc Institute, and TogetherAI, Evo represents a significant advancement in leveraging machine learning for genomic data.

Fig 8. The Evo model architecture, based on StripedHyena. The figure was adapted from https://hazyresearch.stanford.edu/blog/2023-06-29-hyena-dna.
Evo is built on the StripedHyena
architecture, a hybrid model combining 29 layers of data-controlled
convolutional operators with 3 layers of multi-head attention equipped with
rotary position embeddings (RoPE). (Check Fig. 8 and reference about
StripedHyena for detailed
information about the archtecture.) This design enables efficient processing of
long DNA sequences while maintaining single-nucleotide resolution. Evo scales up
to 7B parameters and utilizes a context length of 131 kilobases (kb) at
single-nucleotide resolution. Evo employs byte-level, single-nucleotide
tokenization for input DNA sequences, allowing it to model sequences with fine
granularity. During pretraining, Evo uses an effective vocabulary of four tokens
(A, T, C, G) from a total vocabulary of 512 characters. The additional 508
characters beyond ATCG enable prompting with special tokens during generation
with finetuned models. Meanwhile, RoPE aids in maintaining positional
information over long contexts, crucial for genomic sequences where the relative
position of nucleotides can impact biological function.
Evo was pretrained on a vast dataset of 300 billion nucleotide tokens, encompassing 2.7 million prokaryotic and phage genomes from databases such as the Genome Taxonomy Database (GTDB), Integrated Microbial Genomes/Virus (IMG/VR), and Integrated Microbial Genomes/Plasmid (IMG/PR). The authors introduced OpenGenome in this work, compiling over 80,000 bacterial and archaeal genomes, and millions of predicted prokaryotic phage and plasmid sequences. The training followed a two-stage process: an initial phase with an 8k token context length, followed by a context extension phase to 131k tokens. Evo was trained using an autoregressive modeling approach, predicting the likelihood of the next token given a sequence of tokens. This next-token prediction task was purely on raw genome sequences with no explicit supervision or annotations, which is fundamental for learning how to capture complex patterns in DNA sequences. Evo employs a decoder-only framework, enabling it to efficiently handle long context lengths and maintain single-nucleotide resolution.

Fig 9. Evo models the fundamental modalities of biology as seen in the centra dogma of molecular biology. The figure was from https://arcinstitute.org/news/blog/evo, which is also Fig. 1a from Evo’s paper.
Evo supports RNA and protein sequence modeling through its DNA outputs by leveraging the central dogma of molecular biology (Fig. 9). By modeling genome sequences at single-nucleotide resolution, Evo captures the information encoded in regulatory DNA, protein-coding DNA, coding RNA, and non-coding RNA, etc. It can learn covariation involving multiple genes and regulatory elements, making DNA a productive modality for developing a biological foundation model. However, it is important to note that Evo’s tokenization approach focuses on DNA sequences, and while it can model RNA-related functionality through their DNA counterparts, it does not directly accept RNA sequences as inputs. The training data primarily consists of DNA sequences, and any RNA-related functionality is derived from the model’s understanding of DNA sequences. This approach is also applied to protein sequences.
To support structure-related predictions, Evo’s generated DNA sequences can be processed using tools like ESMFold, AlphaFold2, and RNAMultiFold. These tools help predict the 3D structures of proteins and RNA sequences that Evo generates through its DNA-based learning.
Evo excels in various applications:
- Zero-shot Function Prediction: Evo outperforms domain-specific models (e.g. ESMs, NTs, RNA-FM) in predicting the effects of mutations on protein and ncRNA functions without task-specific finetuning. This capability is derived from its understanding of the DNA sequences encoding these molecules.
- Generative Design: Evo can generate synthetic CRISPR-Cas systems, including coherent protein and non-coding RNA sequences. This is achieved by generating DNA sequences that code for these molecules, which can then be transcribed and translated into functional RNA and proteins.
- Gene Essentiality Prediction: Using long genomic contexts, Evo accurately predicts essential genes in bacteria and phages. By analyzing the DNA sequences, Evo can determine which genes are crucial for an organism’s survival, providing insights into organismal fitness and potential targets for drug discovery.
- Whole-genome Sequence Generation: Evo generates sequences up to 650 kb, demonstrating high coding density and plausible genomic organization. These sequences can include coding regions for proteins and RNAs, enabling further studies on their structure and function using tools like ESMFold, AlphaFold2, and RNAMultiFold.
The authors conducted a comprehensive scaling laws analysis, comparing over 300 models across four architectures: Transformer++, Mamba, Hyena, and StripedHyena. They found that StripedHyena demonstrates favorable scaling laws on DNA sequence data, outperforming other architectures, including state-of-the-art Transformers and modern data-controlled state-space models like Mamba. However, training Evo required substantial computational resources, including 64 Nvidia H100 GPUs and 128 Nvidia A100 GPUs. It was trained on ~340B tokens using ~$2 \times 10^{22}$ FLOPS.
Evo’s primary advantage lies in its ability to handle long genomic sequences at single-nucleotide resolution, enabling comprehensive modeling of biological systems. Its hybrid architecture ensures efficient processing and scalability, outperforming traditional Transformer models on genomic data. Currently trained exclusively on prokaryotic data, Evo’s predictions for eukaryotic sequences, including human genomes, remain limited. Expanding Evo to include eukaryotic genomes will require addressing the much higher complexity of these genomes, along with significant investments in engineering, computational resources, and safety-related model alignment. While Evo is capable of generating broad genomic organization, further improvements are needed in the detail and completeness of generated sequences. Additionally, the model’s substantial computational demands during both training and inference could limit accessibility for smaller research groups.
Methylation-awared Language Models
MethylBert
MethylBERT (Jeong, et al. 2023) is a deep learning model designed for read-level DNA methylation pattern classification and tumour purity estimation. Built on the DNABert architecture, it processes overlapping 3-mer DNA sequences (comprising 69 unique tokens, including DNA bases A, T, C, G and special tokens like [MASK], [UNK]) combined with methylation states to encode read-level methylomes. Unlike traditional methods that rely on microarray-based beta-values, MethylBERT directly analyzes sequencing-based methylation data, preserving single-molecule signals and improving sensitivity for detecting rare cell types. The model’s primary goal is to classify sequencing reads as either tumor or normal cells, facilitating tumor fraction estimation in bulk DNA methylation samples.
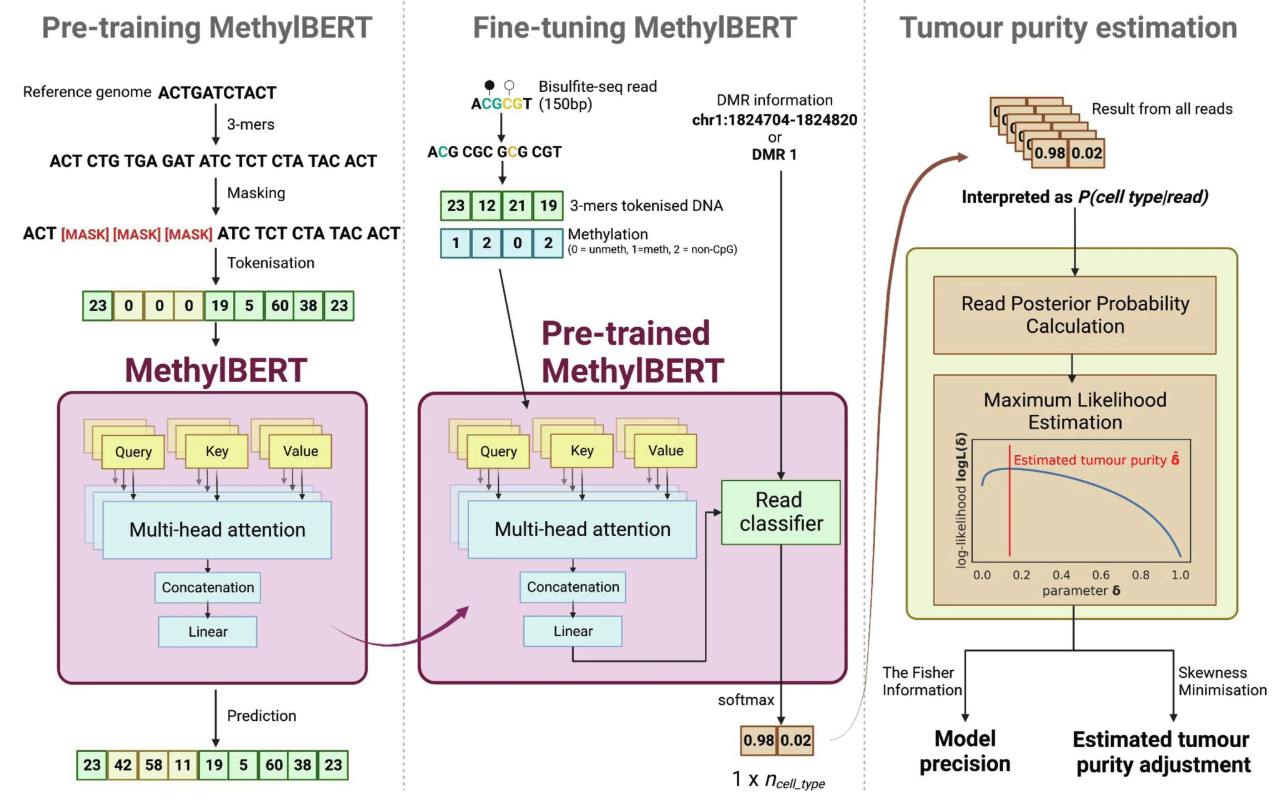
Fig 10. Overview of MethylBERT. The figure is Fig. 1 from its paper.
MethylBERT’s training process consists of two major stages (Fig. 10). During the pre-training stage, the model employs an unsupervised Masked Language Model (MLM) objective on the hg19 genome (or mouse mm10 for cross-species applications). It learns to predict missing 3-mer DNA sequences using contextual embeddings, processing DNA sequences in 510 bp segments. Through this process, the model develops a deep understanding of sequence dependencies, CpG distributions, and genome-wide structures.
In the fine-tuning stage, MethylBERT focuses on specialized read-level methylation classification using supervised learning with tissue-derived data, such as Diffuse Large B-cell Lymphoma and Non-neoplastic B-cells. The model classifies reads based on CpG methylation patterns and genomic context, processing methylation states in a categorical format where 0 represents unmethylated CpG (unmodified cytosine), 1 indicates methylated CpG (cytosine with methyl group), and 2 denotes non-CpG sites (other nucleotides, not used in classification). The model converts these methylation states into embedding vectors and combines them with DNA sequence embeddings for comprehensive representation.
For final predictions, MethylBERT follows a three-step process (Fig. 10): first, it classifies each read as tumor-derived or normal using model embeddings; second, it incorporates additional genomic region information such as DMR labels; and finally, it estimates tumor purity in bulk samples using Bayesian probability inversion and maximum likelihood estimation.
The model’s architecture mirrors DNABert, featuring 12 Transformer encoder layers with 768 hidden units and 12 attention heads. Additionally, the authors developed a smaller variant with 6 encoder layers optimized for longer read sequences (500 bp), which achieved comparable performance to the larger model.
The computational requirements for MethylBERT varied significantly between training stages. The pre-training phase required several days using 4 NVIDIA V100 GPUs, while the subsequent fine-tuning phase completed much more quickly.
MethylBERT exhibited remarkable capabilities in analyzing complex methylation patterns, significantly outperforming traditional statistical approaches like CancerDetector and Hidden Markov Models (HMM). Its particular strength lies in read-level DNA methylation analysis, demonstrating high accuracy and sensitivity. These results highlight MethylBERT’s potential as a powerful tool for liquid biopsy applications, where precise methylation pattern detection is crucial for early disease detection and monitoring.
MethylGPT
Unlike MethylBert, which is a BERT-like encoder model that embeds full sequences with methylation status, MethylGPT is a decoder-only generative model designed to analyze DNA methylation patterns across large numbers of CpG sites (Ying, et al. 2024). The model was trained on a large-scale dataset of 154,063 human methylation profiles, capturing 49,156 physiologically relevant CpG sites and processing a total of 7.6 billion training tokens. Unlike conventional linear models, MethylGPT leverages deep learning to recognize both local and higher-order genomic features, enabling robust predictions in various epigenetic applications.
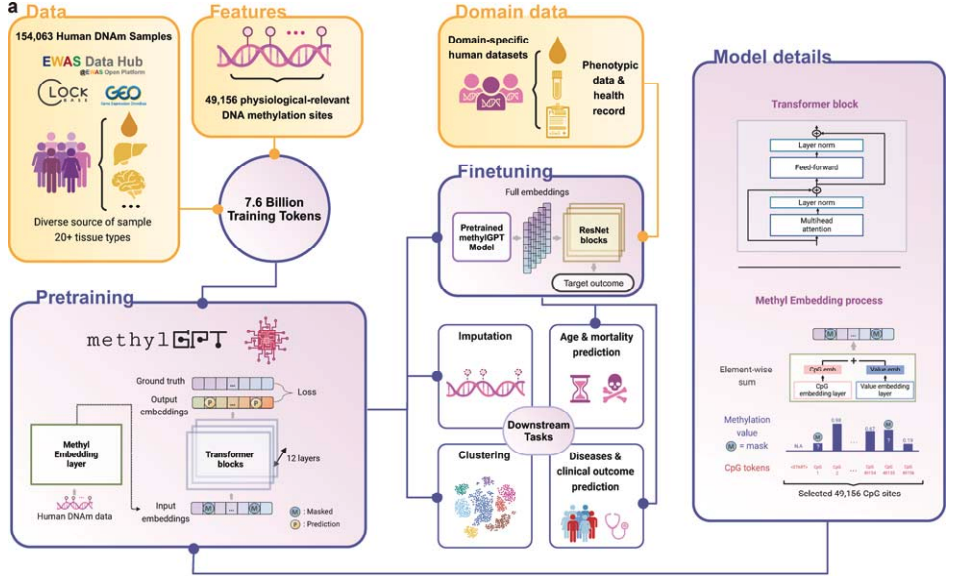
Fig 11. Overview of MethylGPT. The figure is Fig. 1a from its paper.
MethylGPT employs a transformer architecture consisting of six transformer blocks, with each block containing four multi-head self-attention layers followed by a feed-forward network. The model’s tokenization strategy involves two key components: CpG site positions and methylation states. Each CpG site is assigned a unique integer identifier for position encoding, while methylation states are embedded separately. These two types of embeddings are combined through an element-wise operation before being processed by the transformer layers. This dual embedding approach enables the model to simultaneously capture both the genomic context of CpG sites and their methylation states through the attention mechanism. The model uses a 64-dimensional embedding space for representation learning, and includes a special [CLS] token at the beginning of each sequence to generate sample-level representations for downstream tasks.
During pretraining, the model was optimized using two complementary loss functions:
- Masked Language Modeling (MLM): Predicts methylation levels for 30% randomly masked CpG sites.
- Profile Reconstruction Loss: Uses the [CLS] token to reconstruct full DNA methylation profiles.
The training dataset was curated from the EWAS Data Hub and Clockbase, covering over 20 tissue types. The selection of 49,156 CpG sites was based on their association with Epigenome-Wide Association Study (EWAS) traits and their prevalence across datasets. Methylation values were normalized, and missing values were handled through masking during training.
The training process proceeded for 10 epochs using the AdamW optimizer, starting with a 0.001 learning rate and implementing a 10% per-epoch decay. The implementation utilized NVIDIA A100 GPUs with a batch size of 16 and incorporated FlashAttention for efficient memory usage.
MethylGPT was shown to generalizes well across epigenetic tasks, including methylation imputation, age prediction, missing data handling, and intervention analysis. It excels in biological interpretability, scalability, and resilience to missing data, outperforming traditional models in accuracy. However, its high computational cost and training on bulk methylation data may limit generalization to single-cell methylation data. Furthermore, the model only considers CpG sites rather than incorporating the full sequence context, which may limit its ability to efficiently capture sequence-dependent patterns during both training and inference.
RNA Language Models
RiNALMo
RiNALMo (RiboNucleic Acid Language Model) (Penić, et al. 2024) is a RNA language model designed to understand and predict RNA structures and functions. It employs a BERT-style Transformer encoder architecture, which consists of 33 Transformer blocks with an embedding dimension of 1280. Each block includes multi-head attention (20 heads) and feed-forward networks, utilizing advanced techniques such as rotary positional embeddings (RoPE) and the SwiGLU activation function. RiNALMo has a total of 650M parameters.
The model tokenizes RNA sequences by treating each nucleotide as a single token. During preprocessing, all instances of uracil (U) in the sequences are replaced with thymine (T). The resulting vocabulary includes primary nucleotides (A, C, G, T) and various ambiguous nucleotide combinations and special tokens. These ambiguous combinations cover multiple nucleotide possibilities and are used to handle sequences where the exact nucleotide is not known or where multiple nucleotides are possible due to sequencing uncertainties or biological variability:
| Symbol | Nucleotides Represented |
|---|---|
| R | A or G |
| Y | C or U |
| K | G or U |
| M | A or C |
| S | G or C |
| W | A or U |
| B | C, G, or U |
| D | A, G, or U |
| H | A, C, or U |
| V | A, C, or G |
| N | Any nucleotide (A, C, G, U) |
Additional special tokens used in the model include [CLS], [EOS], [PAD], and [MASK]. The positional information of tokens is encoded using RoPE, which captures both relative and absolute positional data, enhancing the model’s ability to learn sequence relationships effectively.
RiNALMo was pre-trained on a dataset of 36 million non-coding RNA (ncRNA) sequences curated from multiple sources including RNAcentral, Rfam, and Ensembl. The model’s pre-training involved a masked language modeling (MLM) task, where 15% of the tokens in the input sequences were masked and the model was trained to predict these masked tokens. This approach helps the model learn the underlying structure and function of RNA sequences from vast amounts of unannotated data.
The pre-training of RiNALMo was conducted over six epochs on a cluster of seven A100 GPUs, each with 80 GB of memory. The training utilized a batch size of 192 per GPU and employed a cosine annealing learning rate schedule with a linear warm-up. The training process involved intensive computational resources to handle the large-scale data and model parameters efficiently.
RiNALMo has demonstrated good performance across various RNA-related tasks. Key applications include:
-
Secondary Structure Prediction: RiNALMo’s embeddings were fine-tuned for predicting RNA secondary structures, exhibiting superior generalization capabilities compared to existing deep learning methods. It was able to generalize well on unseen RNA families, a significant advancement over traditional models.
-
Splice-Site Prediction: The model was fine-tuned to predict splice sites in RNA sequences, outperforming other RNA language models (e.g. RNA-FM) and established methods. RiNALMo’s ability to generalize across different species’ RNA sequences underscores its robustness and versatility.
-
Mean Ribosome Loading (MRL) Prediction: RiNALMo was also fine-tuned to predict MRL values for mRNA sequences, showing superior performance and generalization on human UTRs despite being trained on random sequences.
RiNALMo’s primary advantage lies in its large-scale pre-training on diverse RNA sequences, which equips it with powerful representations that can be effectively utilized across various downstream tasks.
While RiNALMo excels in secondary structure prediction, it struggles with specific RNA families, such as telomerase RNAs. Overall, RiNALMo is a RNA embedding modeling, providing a powerful tool for RNA structure and function prediction tasks. Its ability to generalize across different RNA families and its superior performance on a range of downstream tasks highlight its potential to drive forward our understanding of RNA biology and its applications in biomedical research.
RNAErnie
RNAErnie (Wang, et al. 2024) is a RNA language model that integrates biological knowledge through RNA motifs (Fig. 11). The model employs a transformer-based architecture consisting of 12 layers of multihead transformer blocks, with each layer maintaining a hidden state dimension of 768. At its core, RNAErnie builds upon the Enhanced Representation through Knowledge Integration (ERNIE) framework, which systematically incorporates external domain knowledge during the pretraining process. This knowledge integration enables the model to capture and represent intricate biological relationships within RNA sequences more effectively. Specifically, RNAErnie leverages RNA motifs and RNA type information as biological priors, significantly enhancing its capabilities across various RNA analysis tasks.
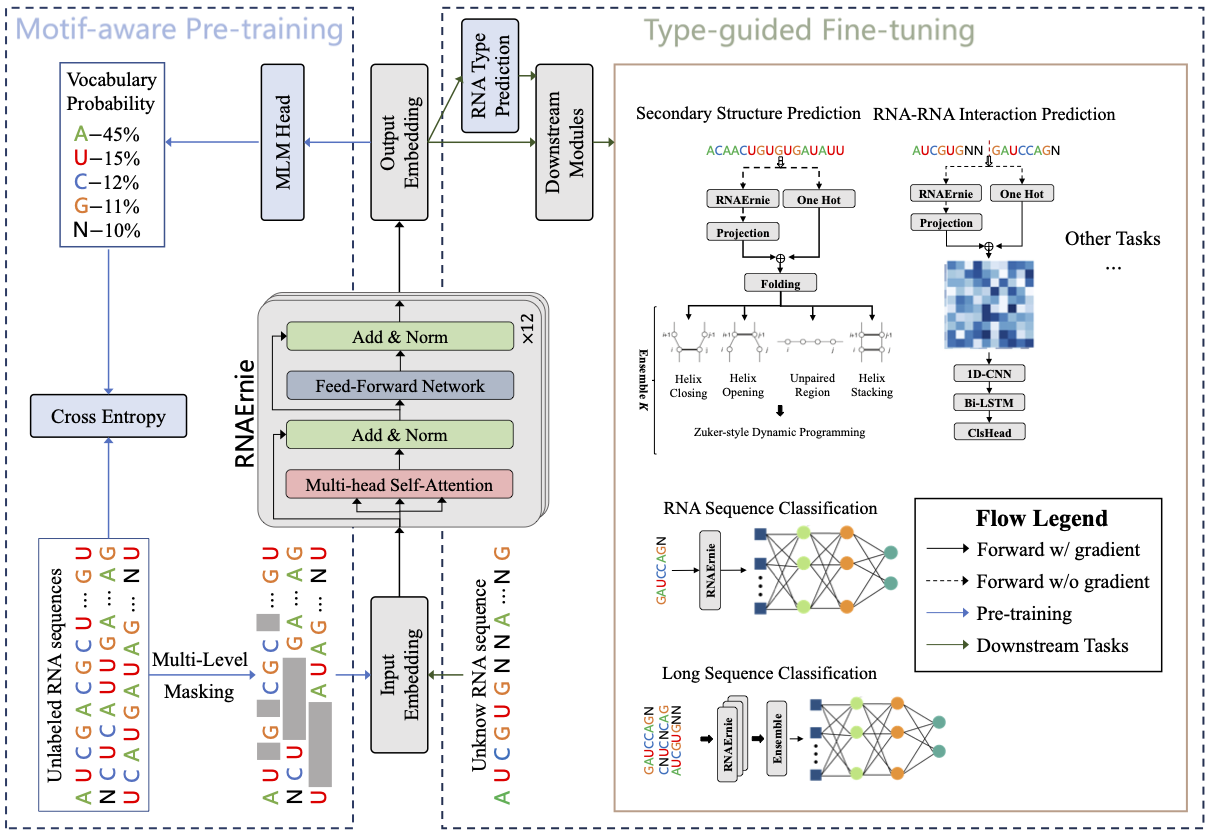
Fig 11.Illustration of RNAErnie. The model uses 12 transformer encoder layers and employs motif-aware pretraining on 23M RNAcentral sequences. It then performs type-guided fine-tuning by predicting RNA types and using them as auxiliary information for downstream tasks. Fig. 1 from original paper.
During pretraining, RNAErnie employs a sophisticated motif-aware multilevel masking strategy that incorporates biological knowledge (Fig. 12a). This hierarchical masking approach consists of:
- Base-level Masking: Randomly masks 15% of nucleobases within an RNA sequence, aiding in learning fundamental token representations.
- Subsequence-level Masking: Masks contiguous segments of nucleobases, ranging from 4 to 8 bases, to capture deeper biological information.
- Motif-level Masking: Incorporates biologically significant motifs from databases like ATtRACT and SpliceAid, masking these motifs to embed complex structural and functional elements within the model.
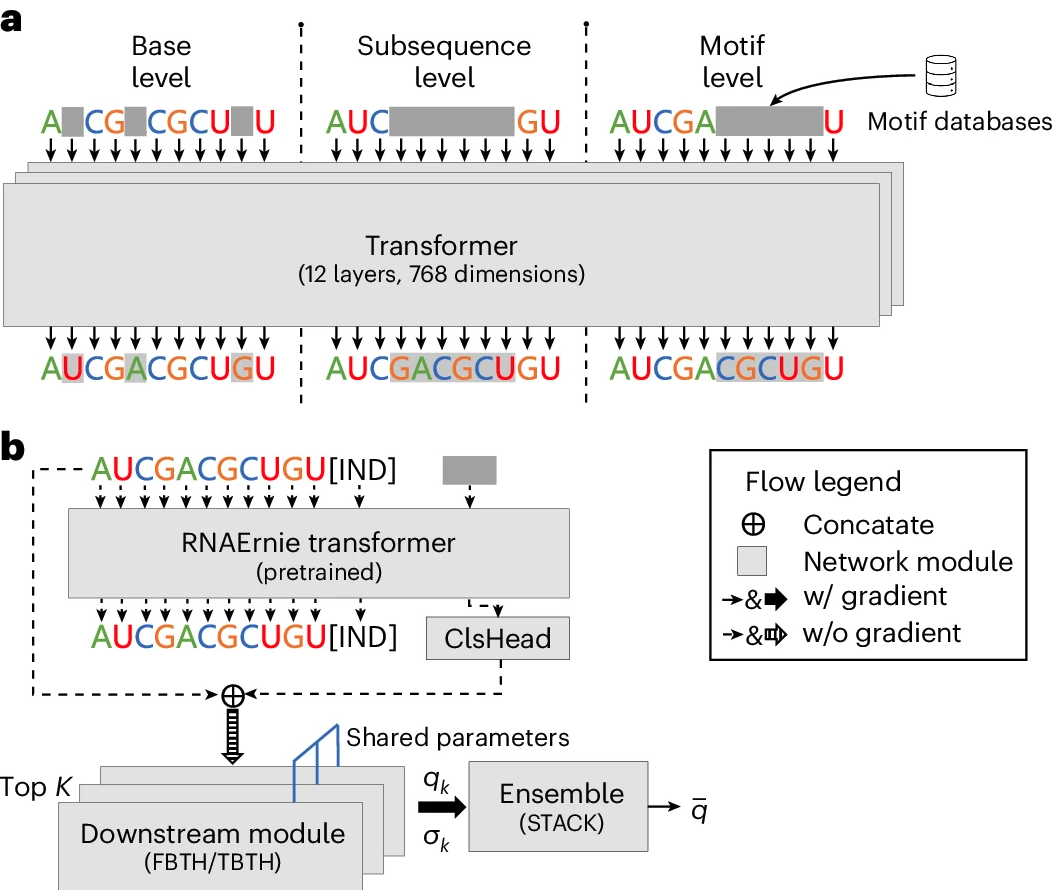
Fig 12. RNAErnie’s training strategies. a, Motif-aware masking with three levels (base, subsequence, motif) during pretraining. b, Type-guided fine-tuning predicts RNA types first, then uses ensemble learning with shared parameters for downstream tasks.
RNAErnie utilizes a type-guided fine-tuning approach, leveraging predicted RNA types as auxiliary information (Fig. 12b). This strategy employs three neural architectures:
- FBTH (Frozen Backbone with Trainable Head): Extracts embeddings from the pretrained RNAErnie block and uses them to train a separate task-specific head.
- TBTH (Trainable Backbone with Trainable Head): Combines the RNAErnie block with task-specific heads into an end-to-end neural network for supervised learning tasks.
- STACK: Uses the RNAErnie block to predict the top-K possible RNA types, followed by ensemble learning through multiple downstream modules.
RNAErnie has approximately 105M trainable parameters and processes RNA sequences by tokenizing the bases ‘A’, ‘U’, ‘C’, and ‘G’. Each sequence is appended with an initial classification embedding ([CLS]) and an indication embedding ([IND]), which helps cluster similar RNA sequences in a latent space for more effective retrieval-based learning. The model accepts input sequences up to 512 nucleotides in length, though this limitation may pose challenges when analyzing complex three-dimensional RNA structural motifs.
The model was pretrained on approximately 23 million RNA sequences from RNAcentral. Training was conducted on four Nvidia Tesla V100 32GB GPUs over approximately 250 hours, using the AdamW optimizer with a learning rate scheduler that incorporates anneal warm-up and decay techniques. The initial learning rate was set to 1×10^-4.
RNAErnie demonstrates robust performance across various RNA analytical tasks in both supervised and unsupervised learning scenarios. The model has been successfully evaluated on RNA sequence classification, RNA-RNA interaction prediction, and RNA secondary structure prediction tasks. Its effectiveness is particularly evident in RNA sequence classification on the nRC dataset, where RNAErnie+ achieved an impressive 96.88% accuracy, significantly outperforming baseline models (e.g. RNABERT, RNA-MSM and RNA-FM).
HELM
HELM (Hierarchical Encoding for mRNA Language Modeling) (Yazdani-Jahromi, et al. 2024) is a new LLM pretraining method specifically designed for analyzing messenger RNA (mRNA) sequences. HELM introduces a pretraining strategy that explicitly incorporates the biological hierarchy of mRNA sequences, particularly at the codon level. This approach better reflects the inherent structure of genetic information, where three nucleotides form a codon that codes for a specific amino acid. By aligning the model architecture and training process with these fundamental biological principles, HELM aims to achieve more accurate and biologically meaningful sequence analysis.
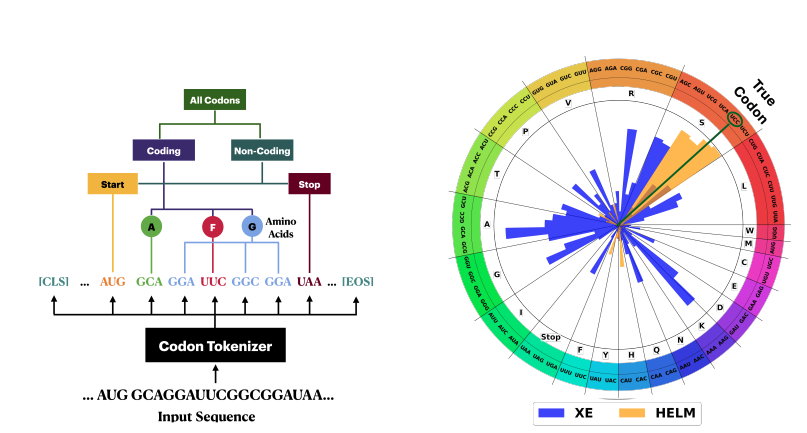
Fig 13.Hierarchical codon-aware tokenization and loss function in HELM. Left: The hierarchical structure of codons used in HELM for tokenization and modeling. Codons are categorized into Start, Coding (grouped by amino acids), and Stop codons. This biologically informed hierarchy influences the training loss function by prioritizing synonymous relationships between codons. Right: Codon prediction probabilities visualized on an amino acid codon wheel. Orange bars represent HELM’s Hierarchical Cross-Entropy (HXE) loss, while blue bars correspond to the standard Cross-Entropy (XE) loss. HELM assigns higher probabilities to synonymous codons when making predictions, better capturing biological redundancy, whereas XE tends to misassign probability to non-synonymous codons.
HELM proposes to adopt Hierarchical Cross-Entropy Loss (HXE, see eq1) to better capture synonymous codon relationships.
\begin{equation} \begin{aligned} L_{HXE} = -\sum_{i} w_i \cdot y_i \log \hat{y}_i \end{aligned} \end{equation}
where:
- $y_i$ is the true one-hot label,
- $\hat{y}_i$ is the predicted probability for codon $i$,
- $w_i$ is a weight assigned based on codon similarity based on $\lambda (C) = exp(-\alpha h(C))$, and $h(C)$ is the height of the node C in the hierarchy and ($\alpha > 0$).
Unlike standard cross-entropy loss which treats all prediction errors equally, HXE implements a structured hierarchy where prediction errors are penalized differently based on their biological significance within the codon structure. In another words, This encourages the model to prefer synonymous codons over random codon assignments, improving biological plausibility in sequence predictions.
The pretraining dataset consists of 15.3 million curated mRNA sequences from the Observed Antibody Space (OAS) database, which includes sequences from over 80 studies. The model was trained using 8 NVIDIA A100 GPUs over 40 epochs. HELM is implemented using multiple architectures, including Transformer, Mamba, and Hyena models, each with 50M parameters.
Key technical features include:
- Codon-level tokenization strategy (64 codons plus special tokens)
- Hierarchical Cross-Entropy Loss (HXE) for biologically-informed error weighting
- Support for both Masked Language Modeling (MLM) and Causal Language Modeling (CLM) training objectives
HELM has demonstrated superior performance than a few baseline methods (e.g. RNA-FM, SpliceBERT, and CodonBERT) in several critical areas:
- Accurate prediction of mRNA properties including protein expression and thermostability
- Precise annotation of antibody regions
- High-quality mRNA sequence generation
In comparative evaluations, HELM achieved significant improvements:
- 8% performance gain over standard bio-language models
- 2x parameter efficiency compared to state-of-the-art models like RNA-FM and CodonBERT
- Enhanced biological plausibility in generated sequences
- Improved accuracy in property prediction tasks through better capture of codon hierarchies
The model shows particular promise for therapeutic applications, especially in vaccine development and gene therapy. However, authors also acknowledged the following limitations:
- Training in Euclidean space may not optimally capture hierarchical relationships compared to hyperbolic space
- The specialized tokenization and pretraining approaches may present integration challenges with existing bioinformatics pipelines
Overall, HELM demonstrates how incorporating domain-specific biological knowledge into both data preprocessing and model training can significantly enhance LLM performance. By explicitly modeling the hierarchical nature of codons and using biologically-informed loss functions, HELM achieves better results with fewer parameters compared to conventional approaches. This suggests a promising direction for developing more efficient and biologically meaningful language models in genomics.
Gene Language Models
scGPT
scGPT (Cui, et al. 2024) is a generative pre-trained transformer model specifically designed for analyzing single-cell RNA sequencing (scRNA-seq) and multi-omics data (Fig. 14). Built on a decoder-only transformer architecture, scGPT processes gene expression data by treating individual genes as tokens and converting each cell’s expression profile into a sequence. This approach allows the model to capture complex relationships between genes, cells, and tissues, learning intricate biological patterns in a manner analogous to how traditional GPT models learn linguistic structures.
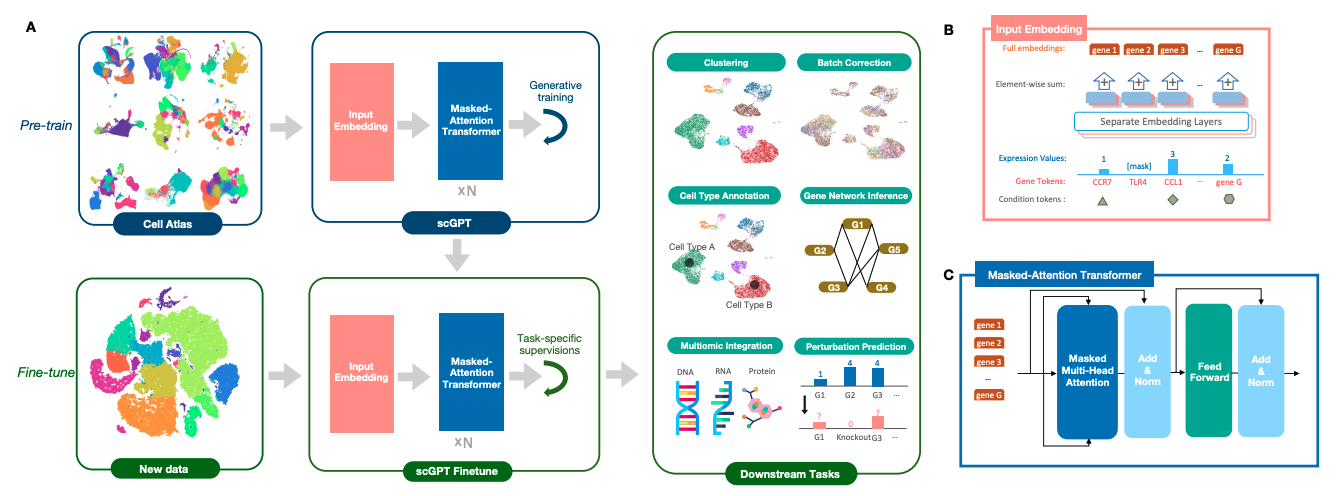
Fig 14. Overview of scGPT.
scGPT employs a sophisticated tokenization strategy:
- Gene names serve as the primary vocabulary tokens
- Expression values undergo pre-defined binning transformations to standardize data across different sequencing modalities
- Condition tokens capture important metadata such as batch identity for technical variation tracking, modality type (e.g., RNA, ATAC-seq, proteomics) and perturbation conditions (e.g., drug treatments, CRISPR knockouts). These condition tokens are crucial for integrating data across different experiments, enabling scGPT to generalize beyond individual datasets and infer biological relationships across diverse conditions.
As a fundatational model, scGPT was pre-trained on an extensive dataset of 33 million single-cell RNA sequencing profiles from the CELLxGENE repository. This dataset encompasses 51 organs and 441 studies, providing broad tissue diversity across healthy human cells. The comprehensive training data enables zero-shot generalization to disease-related tasks, as demonstrated in studies involving multiple sclerosis (MS) and cancer datasets.
The pre-training process utilizes a comprehensive self-supervised learning approach with four main tasks:
-
Masked Gene Prediction: Similar to BERT’s masked language modeling, the model predicts expression values for randomly masked gene tokens based on surrounding context.
-
Gene Expression Imputation: The model learns to reconstruct missing gene expression values, addressing the common challenge of sparse data in single-cell datasets.
-
Modality-Aware Pre-training: Integration of RNA, ATAC-seq, and proteomics data to develop unified multi-omic representations.
-
Batch Effect Reduction: Implementation of contrastive learning techniques to minimize technical variations while preserving biological signals.
The pre-training process leverages high-performance A100 GPU clusters and employs FlashAttention for optimized self-attention computations.
After pre-training, scGPT demonstrates remarkable versatility through fine-tuning for various downstream tasks:
- Cell-type annotation
- Batch effect correction
- Multi-omic data integration
- Perturbation response prediction
- Gene Regulatory Network (GRN) inference
This adaptability makes scGPT a powerful tool for diverse applications in single-cell genomics research. However, like most LLMs, scGPT faces certain limitations like biases in the training data. Since it is pretrained primarily on publicly available single-cell datasets, rare or underrepresented cell types may not be adequately captured in the model’s learned representations. This can potentially impact the model’s performance when analyzing low-abundance cell populations or rare cell states, highlighting the importance of considering dataset bias in model applications.
Citation
If you find this post helpful and are interested in referencing it in your write-up, you can cite it as
Xiao, Jiajie. (May 2023). Biomedical LLMs (2): Genomics. JX’s log. Available at: https://jiajiexiao.github.io/posts/2024-05-12_biollm_genomics/.
or add the following to your BibTeX file.
@article{xiao2024_biollm_genomics,
title = "Biomedical LLMs (2): Genomics",
author = "Xiao, Jiajie",
journal = "JX's log",
year = "2024",
month = "May",
url = "https://jiajiexiao.github.io/posts/2024-05-12_biollm_genomics/"
}
References
-
Ji, Y., Zhou, Z., Liu, H., & Davuluri, R. V. (2021). DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics, 37(15), 2112-2120.
-
Zhou, Z., Ji, Y., Li, W., Dutta, P., Davuluri, R., & Liu, H. (2023). Dnabert-2: Efficient foundation model and benchmark for multi-species genome. arXiv preprint arXiv:2306.15006.
-
Kudo, T., & Richardson, J. (2018). Sentencepiece: A simple and language independent subword tokenizer and detokenizer for neural text processing. arXiv preprint arXiv:1808.06226.
-
Sennrich, R., Haddow, B., & Birch, A. (2015). Neural machine translation of rare words with subword units. arXiv preprint arXiv:1508.07909.
-
Press, O., Smith, N. A., & Lewis, M. (2021). Train short, test long: Attention with linear biases enables input length extrapolation. arXiv preprint arXiv:2108.12409.
-
Dao, T., Fu, D. Y., Ermon, S., Rudra, A., & Ré, C. F. fast and memory-efficient exact attention with IO-awareness. arXiv; 2022. arXiv preprint arXiv:2205.14135.
-
Dalla-Torre, H., Gonzalez, L., Mendoza-Revilla, J., Carranza, N. L., Grzywaczewski, A. H., Oteri, F., … & Pierrot, T. (2023). The nucleotide transformer: Building and evaluating robust foundation models for human genomics. BioRxiv, 2023-01.
-
Su, J., Ahmed, M., Lu, Y., Pan, S., Bo, W., & Liu, Y. (2024). Roformer: Enhanced transformer with rotary position embedding. Neurocomputing, 568, 127063.
-
Shazeer, N. (2020). Glu variants improve transformer. arXiv preprint arXiv:2002.05202.
-
Kaplan, J., McCandlish, S., Henighan, T., Brown, T. B., Chess, B., Child, R., … & Amodei, D. (2020). Scaling laws for neural language models. arXiv preprint arXiv:2001.08361.
-
Avsec, Ž., Agarwal, V., Visentin, D., Ledsam, J. R., Grabska-Barwinska, A., Taylor, K. R., … & Kelley, D. R. (2021). Effective gene expression prediction from sequence by integrating long-range interactions. Nature methods, 18(10), 1196-1203.
-
Benegas, G., Batra, S. S., & Song, Y. S. (2023). DNA language models are powerful predictors of genome-wide variant effects. Proceedings of the National Academy of Sciences, 120(44), e2311219120.
-
Nguyen, E., Poli, M., Faizi, M., Thomas, A., Wornow, M., Birch-Sykes, C., … & Baccus, S. (2024). Hyenadna: Long-range genomic sequence modeling at single nucleotide resolution. Advances in neural information processing systems, 36.
-
Jeong, Y., Gerhäuser, C., Sauter, G., Schlomm, T., Rohr, K., & Lutsik, P. (2023). MethylBERT: A Transformer-based model for read-level DNA methylation pattern identification and tumour deconvolution. bioRxiv, 2023-10.
-
Ying, K., Song, J., Cui, H., Zhang, Y., Li, S., Chen, X., … & Gladyshev, V. N. (2024). MethylGPT: a foundation model for the DNA methylome. bioRxiv, 2024-10.
-
Zvyagin, M., Brace, A., Hippe, K., Deng, Y., Zhang, B., Bohorquez, C. O., … & Ramanathan, A. (2023). GenSLMs: Genome-scale language models reveal SARS-CoV-2 evolutionary dynamics. The International Journal of High Performance Computing Applications, 37(6), 683-705.
-
Penić, R. J., Vlašić, T., Huber, R. G., Wan, Y., & Šikić, M. (2024). Rinalmo: General-purpose rna language models can generalize well on structure prediction tasks. arXiv preprint arXiv:2403.00043.
-
Wang, N., Bian, J., Li, Y., Li, X., Mumtaz, S., Kong, L., & Xiong, H. (2024). Multi-purpose RNA language modelling with motif-aware pretraining and type-guided fine-tuning. Nature Machine Intelligence, 1-10.
-
Yazdani-Jahromi, M., Prakash, M., Mansi, T., Moskalev, A., & Liao, R. (2024). HELM: Hierarchical Encoding for mRNA Language Modeling. arXiv preprint arXiv:2410.12459.
-
Cui, H., Wang, C., Maan, H., Pang, K., Luo, F., Duan, N., & Wang, B. (2024). scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nature Methods, 1-11.
-
SentencePiece is a tokenization method that segments text into smaller units such as subwords or characters, allowing for efficient handling of various languages and scripts. It employs techniques like Byte-Pair Encoding (BPE) or unigram language model to create a unified vocabulary, facilitating better representation of rare or out-of-vocabulary words. ↩︎
-
Byte-Pair Encoding (BPE) tokenization is a data compression algorithm that constructs tokens by iteratively merging the most frequent (statistics from the training corpus) pair of consecutive bytes or characters in a corpus to create new tokens, effectively reducing the vocabulary size while preserving meaningful units. This process continues until a predefined vocabulary size or iteration limit is reached, resulting in a compact representation of the original text data. More reading materials and examples can be found from Huggingface. ↩︎
-
Information leakage could happen if one generates k-mer vocabulary by checking all corpus including test data. ↩︎
-
The DNABERT-2 paper says the input limit for DNABERT is 512bps. ↩︎
-
Each token has 6bps. Therefore, 1k tokens lead to 6kb sequence length. ↩︎
-
RoPE encodes absolute positions using a rotation matrix while incorporating relative position dependencies directly into the self-attention mechanism. This approach allows models to handle sequences of different lengths and gradually reduces the influence of distant tokens. RoPE is especially beneficial for extrapolating to longer sequences than those seen during training and has been widely adopted in modern transformer models. Additionally, It also integrates seamlessly with linear self-attention architectures, enabling efficient positional encoding even in models optimized for large-scale processing of long sequences. ↩︎
-
The Poisson negative log-likelihood loss is a loss function often used when modeling count data, such as predicting the number of occurrences of an event. This loss is particularly suitable for data that follows a Poisson distribution: $P(k|\lambda) = \frac{\lambda^k e^{-\lambda}}{k!}$, where the rate parameter $\lambda$ equals both mean and variance of the distribution. Corresponding loss is $\text{PoissonNLLLoss} = -\log(P(k|\lambda)) = \lambda
- k\log(\lambda) + \log(k!)$, where $k$ is the target and $\lambda$ is the expected count that the model would like to predict. Pytorch has provided a built-in loss for this here.